
 NAVIGATION
NAVIGATION
The race to develop a safe and effective vaccine against COVID-19 is on, and it has everyone talking. This is the second of a three-part series on the fundamentals of immunization.
Part 1 of the series reviewed and clarified fundamental terms that are central to the conversation and provided answers to some of the most commonly asked questions.
Part 2 establishes a solid foundation of the current science and provides insight into the regulatory realities that control a vaccine’s path to market, the impact vaccines have had on our society, and provides a glimpse into the future of vaccine technology.
The third and final segment will look closely at the leading candidates for a safe and effective COVID-19 vaccine.
Q: Who is responsible for assuring the safety and effectiveness of vaccine technology?
A: The FDA’s Center for Biologics Evaluation and Research (CBER) is the government agency responsible for monitoring and approving vaccines developed by the private sector for distribution in the Unites States. CBER works closely with the Centers for Disease Control and Prevention (CDC) and the National Institutes of Health (NIH).
Newly developed vaccines are licensed for sale in the US only after thorough development studies, extensive clinical trials to prove safety and efficacy, and inspection and approval of the production methods and facilities. After approval, long-term safety monitoring continues as long as the product is in use, and for some time after.
Q: Why does it take such a long time to bring a new vaccine to market?
A: Although vaccine technology has been in use for over 200 years, development of a new vaccine is a complicated, laborious and expensive process that requires talent, time, and resources from the private and public sectors.
Each time a new pathogen emerges, scientists must conduct research to seek to find answers to pathogen-specific questions, such as:
- How does the pathogen produce disease?
- Which components of the pathogen are responsible for the symptoms of the disease?
- Which components should be the target of the immune response?
- Can the immunity-producing component(s) of the pathogen be separated from all other components?
- How many types of the pathogen exist, and does the path to immunity differ for each?
- How can a supply of the pathogen be grown and maintained in a laboratory and manufacturing setting so that it can be used for studies and eventual vaccine production?
- What is the safest way to expose patients to the pathogen without exposing them to the actual disease – can a live form of the pathogen be weakened and delivered safely?
- What is the potential usefulness of existing vaccine technologies?
- If a novel technology is required, how long and how expensive will the path to market be?
Once those answers have been found, the next step is to conduct pre-clinical development efforts to plan for production of the most suitable prototype(s) and obtain regulatory permission prior to testing theories on humans. This includes:
- Develop plans for (and produce) prototype vaccine candidates based on the knowledge gained during research.
- Develop animal and laboratory tests needed to test all the theories reached.
- Design and conduct pre-clinical testing on animals.
- File a lengthy and detailed application to conduct studies in humans of an Investigational New Drug (IND) with FDA’s Center for Biologics Evaluation and Research (detailing specifications that cover everything from evidence of product sterility, the approach to 3 distinct phases of clinical evaluation, selection of investigators, and the approach to testing in humans).
- Select and qualify testing locations and study team members.
- Plan and develop detailed plans and protocols for conducting testing on humans.
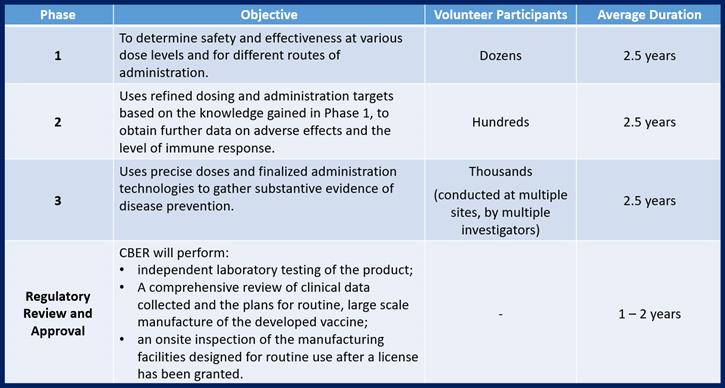
Once governmental approval to conduct studies has been granted, the next step is to conduct 3 phases of safe Clinical Testing on humans to prove all of the theories developed to date. The knowledge gained during each phase of clinical testing is used to refine development efforts before preceding into the next phase of testing. This means that there is additional time spent between each phase to report and interpret the data collected, to make adjustments to the approach that will be taken in the next phase (adjustments in manufacturing the clinical material and in the testing protocols that will be used) and potentially, to make adjustments to the vaccine itself.
In accordance with Federal Regulations, any Phase 1, 2, or 3 study conducted on humans must be conducted by an objective and accredited institution (such as a university or a hospital) and that study must be pre-approved by that institution’s review board. The requirement to use independent (no financial ties to the vaccine developers) facilities, review boards and investigators exist to ensure objectivity during study execution.
Phase 3 studies are generally conducted at more than one institution and by different investigators to further ensure objectivity.
Strict regulations exist to protect the health and safety of all volunteers also exist and apply not only to pre-execution review, but also to general study execution and the active monitoring of ongoing studies.
The licensing/approval process that follows testing the product on humans includes preparation and submission of a lengthy application package to CBER. Once that submission is received, CBER will submit the vaccine for independent testing in an FDA laboratory. They will also review all records of production of the vaccines used in clinical testing, the results of the clinical testing, and the plans established for eventual routine manufacturing and distribution. If all of those review steps are successful, FDA inspectors will perform an on-site inspection of the intended manufacturing facility to verify that the reality is capable of supporting all of the claims made in the plans submitted.
Q: What impact has vaccination had on our society?
A: Serious attempts to immunize individuals and populations began in the 18th century in response to a smallpox epidemic, and since that time the progress made in the global fight against infectious disease has been remarkable.
Vaccination is now a widely used public health tool that has had great success, most notably:
- In 1980 the World Health Organization announced the global eradication of smallpox.
- In the last 40 years, cases (in the United States) of mumps have fallen by 77%, rubella by 96%, pertussis by 97%, diphtheria by 99.7%, and measles and polio by 99.9%,
- U.S. tetanus cases and deaths dropped steadily from a high of 601 cases in 1948 to fewer than 35 cases a year currently.
Q: What are the highest priority goals for the future of immunization efforts?
A: While the efforts to vaccinate has made huge progress protecting us from infectious diseases, there is still progress to be made, such as:
- Reducing the cost of production and distribution of existing vaccines to ensure the densely-populated and poorest areas of the world (where they are needed most) have access to them.
- Researching and developing effective vaccines for diseases that we have not yet conquered (such as HIV/AIDS and malaria).
- Speeding the pathway to safe approval of new vaccines to allow effective response to emerging pandemic situations (such as COVID-19).
Q: What is being done to help us achieve those goals?
A: New vaccine technologies are being actively researched:
- Live Recombinant Vaccine technology uses a weakened pathogen from one disease to deliver an immunogenic protein from another pathogen. HIV research is focusing on this technology because HIV cannot be weakened enough to be safely used in vaccines; the risk that it might cause disease is too high.
- DNA Vaccine technology injects DNA code for a particular antigen, directly into the muscle tissue of a human. The DNA inserts itself into the human’s cells, which then produce the antigen from the infectious agent eliciting an immune response from the body. This technology is relatively easy to produce because DNA is very stable and easy to manufacture. Research and development in this field is ongoing, but as of yet, DNA technology has not yet been shown to yield an effective immune response. However, researchers remain hopeful that DNA vaccine technology may be able to generate immunity against parasitic diseases such as malaria.
In addition to researching new technologies for the vaccines, every effort is being made to improve and simplify the technology used to deliver them. This is needed in order to increase the availability of vaccine technology in areas that lack access to medical professionals or facilities with appropriate refrigeration. Efforts in this area include:
- Wider usage of inhalant delivery systems (nasal sprays).
- Further development of delivery systems that use patches containing a matrix of extremely tiny needles.
- Developing treatments for vaccine materials that would allow temperature sensitive vaccines to be easily transported and stored in a range of environmental (temperature) conditions.
In addition to the technology-focused research that will lead to the development of needed vaccines, and research to improve the efficacy of existing vaccines, valuable efforts are also being made to reduce the lead time needed to safely develop and test novel vaccines.
Part 1 of this series provided a review of the fundamental concepts of the 200 year old effort to eradicate infectious disease. Part 2 provided insight into the regulatory path to market and some of the highest profile objectives of vaccine improvement efforts.
Our third and final segment will evaluate the actions taken to race toward timely development, production, and delivery of an effective vaccine against COVID-19, and what they may mean for our ability to tackle this and future pandemic situations.
The Fundamentals of Immunization, Part 3
The Fundamentals of Immunization, Part 1

Gina Guido-Redden is a quality and regulatory professional with over 25 years of domestic and international industry experience. She is the co-founder and chief operations officer of Coda Corp USA, which provides consultancy services to pharmaceutical, biologics and medical device firms.
Guido-Redden’s history specializes in the areas of facility start up, regulatory compliance and remediation, quality system development, mentorship and training, quality system design, and implementation and management.
She is also a quality systems subject matter expert (SME), frequent seminar presenter, and content contributor to industry publications, including GAMP’s White Paper on Part 11, The Journal of Validation Technology, New Generation Pharmaceuticals, Computer Validation Digest, and MasterControl’s GxP Lifeline. Coda Corp USA is an enterprise partner of MasterControl.
